(Note: it is recommended that you be familiar with the material in the data file conversion section before reading this section.)
LAMARC is a coalescent analysis program. It constructs and analyzes many different possible genealogies (trees) representing the common ancestor relationships among the data sequences sampled. You will need to guide LAMARC's search by specifying how samples from different portions of your organism's genome are related. For example,
In order to guide LAMARC's search through appropriate genealogies, you will need to be able to partition your data samples into
Modeling these different properties will allow you to do an analysis with multiple chromosomal regions and/or multiple populations. You can combine several files together into a single LAMARC input file. These input files do not need to be in the same file format, as long as they are all files the converter can read (i.e. you can stick together two PHYLIP files and a MIGRATE file into a single LAMARC dataset).
Older versions of LAMARC only allowed one to mix different data types only when they were unlinked from each other (i.e. in different linked segment regions). As of LAMARC 2.1, we have relaxed even this restriction, allowing you to mix and match different data types even when they are linked. So, for example, the increasingly-popular data type of microsatellite next to a SNP may now be modeled in LAMARC and will be analyzed appropriately.
A coherent segment (or 'segment' for short) is:
Note that "missing data" is defined differently for different data types. For example, SNPs come as markers and a series of spacings. However, a set of SNPs count as a 'coherent segment' -- while we don't know what the sequence is between the SNPs, we do know that it doesn't vary. Linked microsattelite data is similar -- the number of microsat repeats and their relative locations are relevant, not the sequences between them.
Occasional missing or corrupted data is represented in the input files with a "?" character.
LAMARC's default assumption is that sampled locations within a single segment have the same mutation rate. This assumption can be changed with the rate categories feature in the lamarc menu. However, that feature has LAMARC identify different mutation rates for each site.
When you have sections within a stretch of sampled data for which you know that the mutation rates differ, it may be more appropriate to model them as different coherent segments within the same linked segment region. For example, if you have sequenced a gene with multiple introns and exons, you can include each intron and exon as a coherent segment, each with an appropriate relative mutation rate, and then combine them all into a single linked segement region.
Note: As of LAMARC 2.1, relative mutation rates can only be set from the data model menu of the lamarc program. If you wish to model introns and exons with separate relative mutation rates, place them in separate coherent segments during the conversion process, and set the relative mutation rates from the lamarc program itself. A more complete discussion of the many ways you can accomodate data with different mutation rates is found in section Combining Data with Different Mutation Rates.
Below is a screen shot from the lamarc converter showing the detail panel for coherent segment chrom2-segment1 that results when command file sample-conv-cmd.xml (actual xml is here) is read into the lamarc converter.
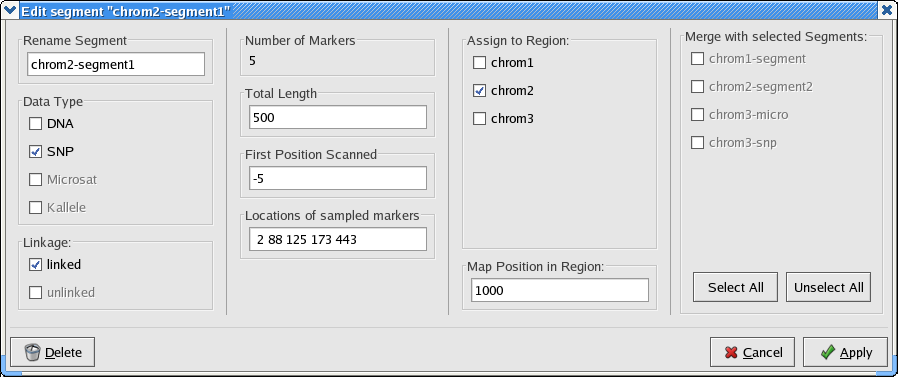
The second column from the left displays the following quantities, which together specify the relative spacing of data within a segment.
The last two of these quantities are measured in "segment coordinates": they are local to the appropriate segment. Thus, if your first position scanned is -5 and your first location is 2, your first SNP is the 7th position scanned. (If you're wondering why it isn't the 8th position, see question Does LAMARC use 'site 0'? Do I?.)
See also Region Coordinates.
Once your data is divided up into coherent segments, if any of these are genetically linked to one another, you can combine them into a linked segment region (or 'region' for short). Unrelated coherent segments each belong in their own region.
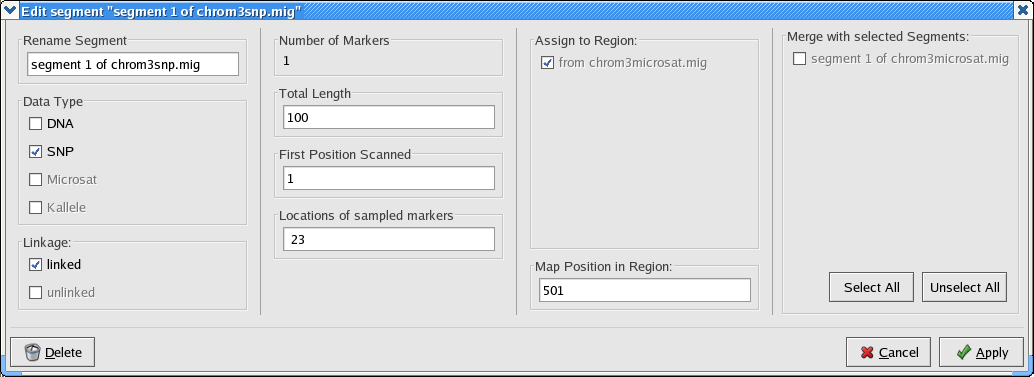
The Segment Panel pictured above for chrom2-segment1 displays a Map Position of 1000. Whenever you have more than one coherent segment in a region, you will need to know their relative spacing. This is entered as the "map position" and is required to:
All Map Positions are given using a single coordinate system for the region, the "regional coordinates" system. If you wish to use region-wide coordinates within segments as well, you may. Your first position scanned would be identical to your map position and all location values should have values between your map position value and the map position plus the length.
Do not put samples together in a region if you have reason to believe they are actually unlinked. This will result in wrong answers. If you put unlinked markers together in a region and also estimate recombination, you will get wrong answers and the program will bog down horribly as it tries to estimate an infinite recombination rate.
The total length of genome that can be included in a single region when estimating recombination has an upper limit of about a centimorgan, though an extensive study with many samples of that length might bog down LAMARC considerably and run very slowly. A more reasonable length is probably half of that. As a quick rule of thumb, we've found that LAMARC runs smoothly for runs where Theta times the recombination rate times the total length of a region is 10 or less.
If you have data with a microsatellite next to a SNP, you want the microsatellite as one coherent segment, the SNP as another coherent segment, and both segments as part of the same linked segment region. This section of the documentation walks you through creating such a file with the converter in GUI mode.
Below is a picture of the converter after you have read in the files chrom3microsat.mig and chrom3snp.mig, and have set the data types appropriately.
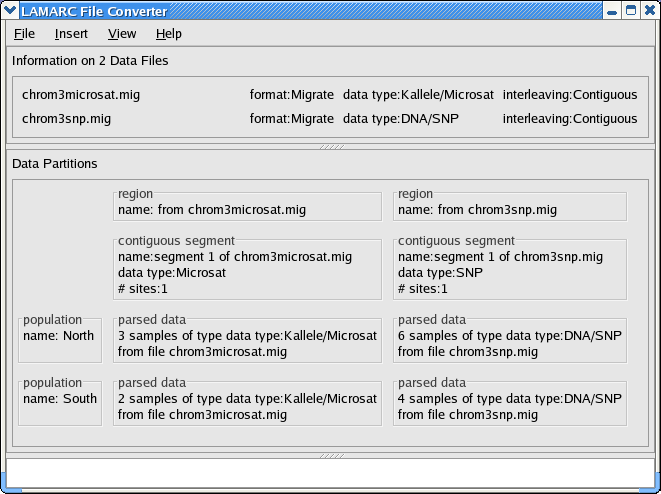
Since we are modeling an adjacent microsat and SNP, we need to place them in the same region. Otherwise, LAMARC will analyze them separatedly. To do this, double click the text inside either of the boxes labeled region within the Data Partitions.
You'll see a panel similar to this:
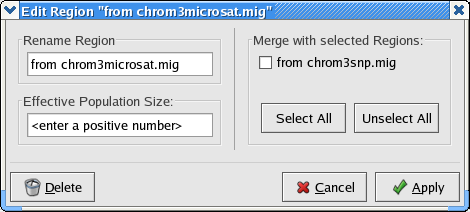
Select the single box within the Merge with selected Region area and click Apply. When you return to the main GUI window, you will notice that the two coherent segments are now included in one region like this:
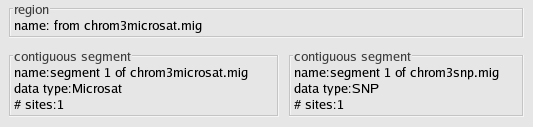
Unfortunately the two data files name their samples in different ways: the SNP file names each haploid sample while the Microsattelite data names each individual. If you try to write a lamarc file at this point, you will get this error:
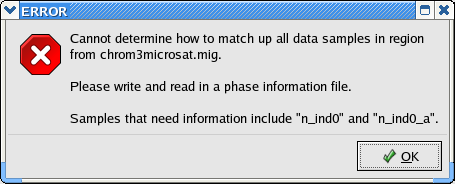
The solution is to write a phase information file like this:
<lamarc-converter-cmd>
<individuals>
<individual>
<name>n_ind0</name>
<sample><name>n_ind0_a</name></sample>
<sample><name>n_ind0_b</name></sample>
</individual>
<individual>
<name>n_ind1</name>
<sample><name>n_ind1_a</name></sample>
<sample><name>n_ind1_b</name></sample>
</individual>
<individual>
<name>n_ind2</name>
<sample><name>n_ind2_a</name></sample>
<sample><name>n_ind2_b</name></sample>
</individual>
<individual>
<name>s_ind0</name>
<sample><name>s_ind0_a</name></sample>
<sample><name>s_ind0_b</name></sample>
</individual>
<individual>
<name>s_ind1</name>
<sample><name>s_ind1_a</name></sample>
<sample><name>s_ind1_b</name></sample>
</individual>
</individuals>
</lamarc-converter-cmd>
You can also find the actual xml
chrom3_phase_cmd.xml here
Read it in using the menu commands File > Read Command File.
Alas, we are still not ready to write a Lamarc file. Attempting to do so results in this error:
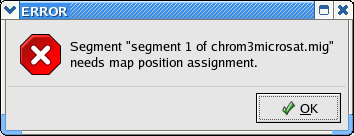
The problem is that we don't know how close together the microsattelite and the SNP are. To solve this problem you'll need to edit fields on the segment panels for each coherent segment.
Begin by setting the map position of the microsatelite segment to 500. Then edit the snp segment as shown here.